高效可再生能源制氢是能源供应从“碳循环”到“氢循环”转变的核心技术之一。然而这一技术仍然面临能量转换效率低这一严峻挑战。能量之间的耦合增益效应是实现能量转换效率最大化的一种潜在途径,但仍需要探索能量耦合增益的物理机制和材料基础。对于电化学水氧化(OER)反应来说,电子是能量转换的载体,只有当体系内能量共同改变电子化学势时才能实现高效的能量耦合增益效应。对于电化学水分解反应来说,需要将电极极化到一个特定能级(催化中心氧化),驱动含氧中间体(*OH,*O,*OOH)中的电子转移到高价活性中心,而后经由催化剂体相转移至外电路。因此,水氧化过电位源自于电解液/催化层/催化剂体相界面处较高电子转移能垒。材料电子态主导了催化活性中心与反应物之间的相互作用,决定了催化反应电子转移势垒。以往研究均以元素掺杂、缺陷态、晶面工程、晶格畸变等直接调控催化材料电子态的方法来降低电子转移势垒。不同于以往直接电子态调控方法,课题组提出了基于材料能量响应特性的能量场调控电子态的方法来调控电子转移势垒 (Adv. Funct. Mater. 2022, 32, 2111234;Nano Lett. 2022, 22, 9131-9137)。
强关联电子体系的电子态取决于电子库伦作用力(U)和电子转移能(∆),比如,Fe基钙钛矿型氧化物的(FeO6)8-八面体具有负电荷转移能,此时由于Fe3d - O2p轨道强相互作用产生配体空穴而使得Fe4+(3d4,t2g3eg1)电子态稳定。然而,在低温下,由于强的p-p轨道电子波动使得配体空穴不稳定而容易诱发Fe4+(3d4,t2g3eg1)电荷歧化2Fe4+(3d4,t2g3eg1)→ Fe3+(3d5,t2g3eg2)+ Fe5+(3d3,t2g3eg0),产生3d轨道被单电子完全占据的Fe3+(3d5,t2g3eg2)(图1)。这意味着,加热至电荷歧化温度(TCD)以上抑制电荷歧化是稳定Fe4+电子态的有效途径。当电荷歧化被热抑制时,体相材料中Fe4+(t2g3eg1)的未占据eg轨道可以作为电子传输通道而无需克服电子翻转或配对能。
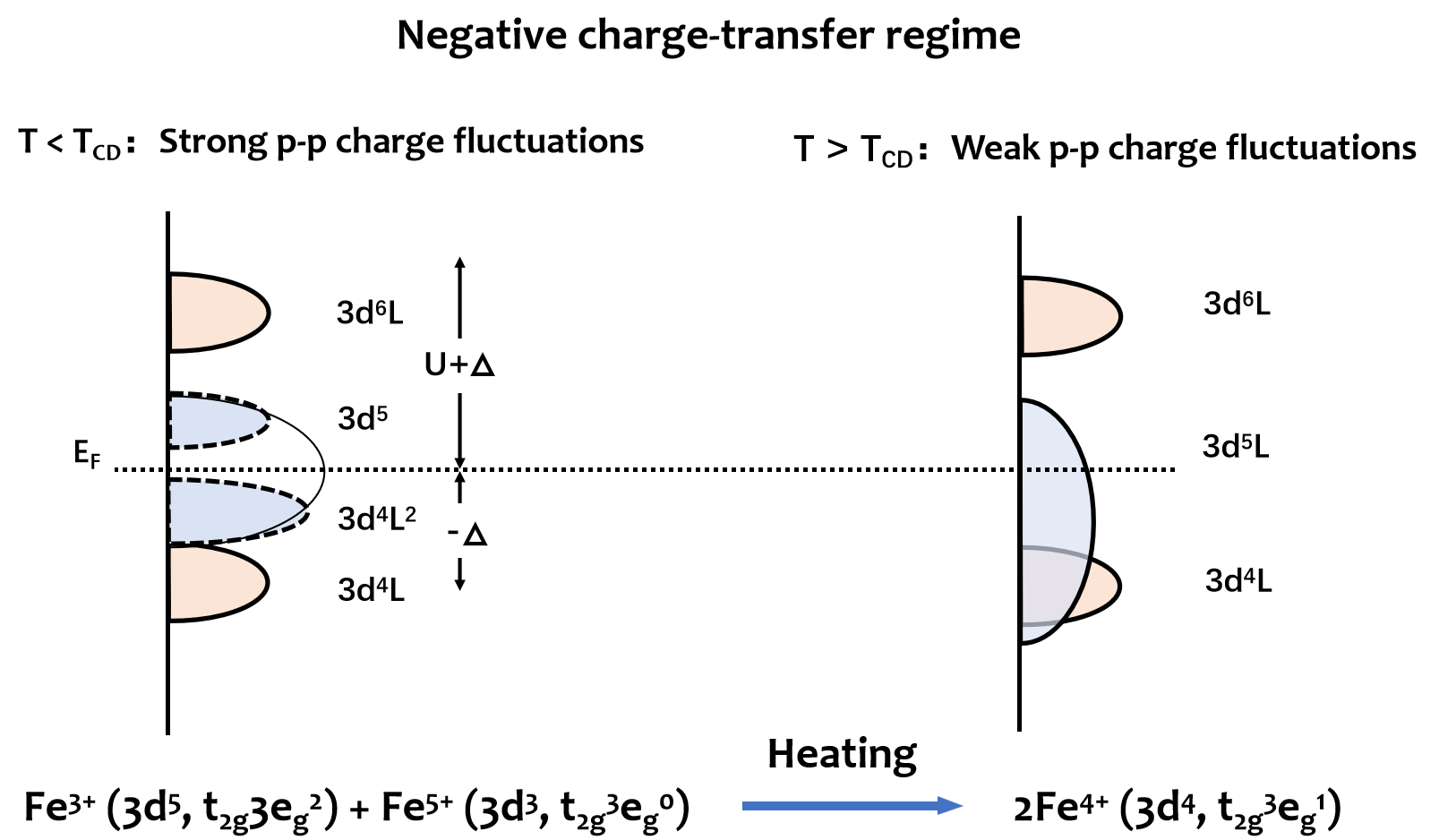
图 1. 负电荷转移机制下Sr3Fe2O7的绝缘体-金属转变的态密度(DOS)和热抑制电荷歧化示意图。黄色和浅蓝色区域分别为Fe 3d和配体O 2p的DOS。虚线曲线表示p-p带隙的打开,U是d-d库仑能相互作用能,∆是配体到金属的电荷转移能,dnL(L:配体空穴)是强电子关联的能态,EF是费米能级。
在此,我们以Ruddlesden-Popper相Sr3Fe2O7钙钛矿氧化物为研究对象,Sr3Fe2O7半导体中的配体空穴在低温下是不稳定的,会引发电荷歧化,并伴随着FeO6结构基元的呼吸振动(图2A, B)。随着温度的升高,比热容导数在67 oC不连续,表明Sr3Fe2O7在此温度点发生了二阶相变(图2C),同时电阻率显著下降(图2D)。场冷和零场冷M-T曲线确认了Sr3Fe2O7反铁磁-顺磁相变奈尔温度为-153 oC,这意味着比热容变化和电阻率变化并不起源于材料磁性转变。根据Curie-Weiss定律,求得Sr3Fe2O7中Fe的有效磁矩为5.25 μB(图2E),说明Fe 3d电子处于高自旋态。Mössbauer谱和变温XPS(图2F,G)则证明了热激励可有效抑制Sr3Fe2O7的电荷歧化,在高于TCD温度,Fe4+电子态稳定。
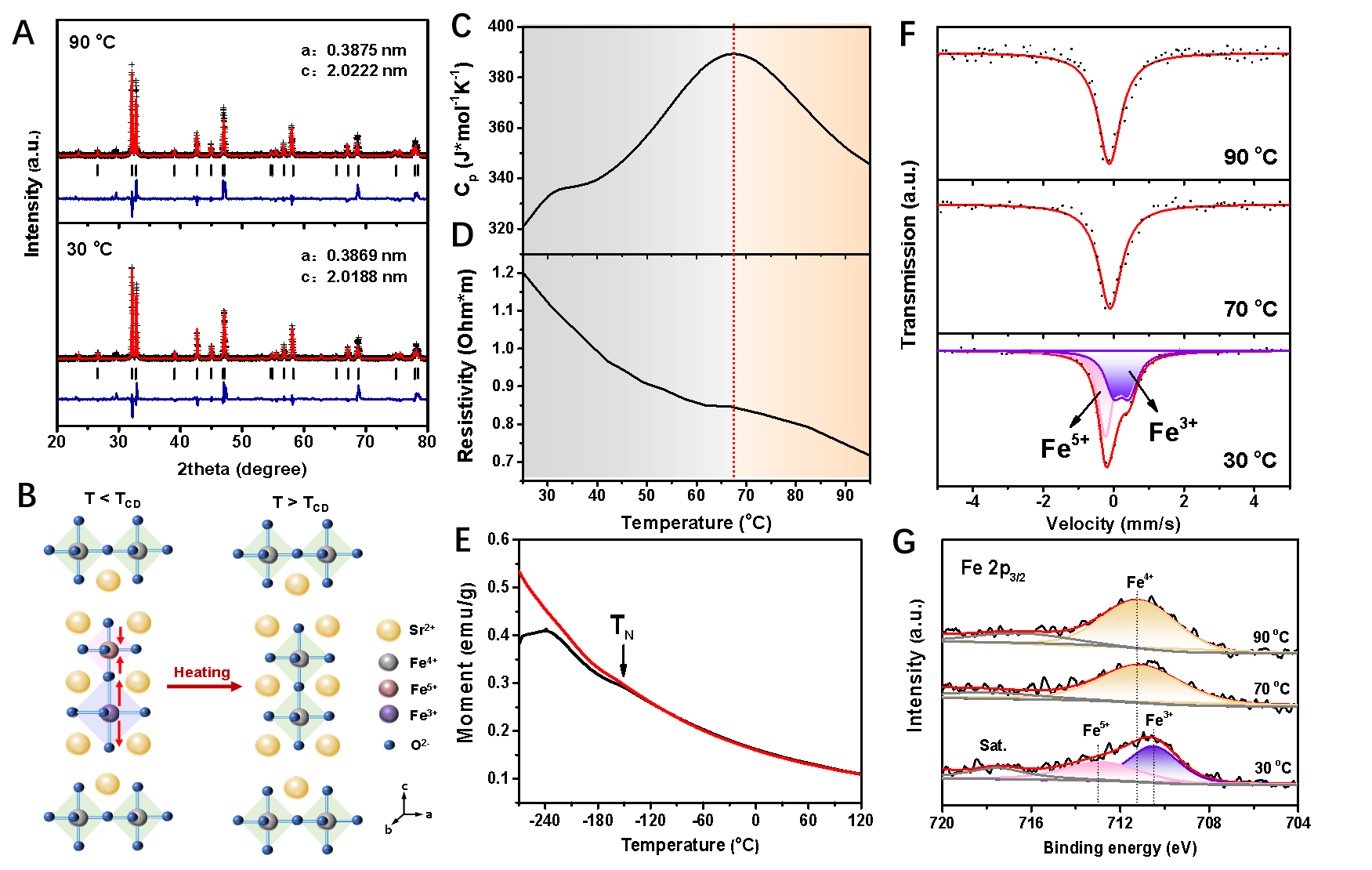
图2.(A)Sr3Fe2O7在30和90 oC下的XRD Rietveld精修。(B)Sr3Fe2O7的四方晶体结构。T < TCD时,Fe4+发生CD并伴随着晶格的呼吸振动;T > TCD时CD和晶格呼吸振动消失。(C)Sr3Fe2O7的比热容随温度的变化。热不连续性表明在TCD = 67 oC处存在二阶相变(虚线)。(D)温度依赖的电阻率。(E)Sr3Fe2O7的零场冷却(ZFC,黑线)和场冷却(FC,红线)磁化曲线。(F)顺磁Sr3Fe2O7在30、70和90 oC时的57Fe Mössbauer谱及其拟合谱。(G)Sr3Fe2O7在30、70和90 oC时的原位Fe 2p XPS光谱。

图3.(A,B)在90 oC的1.0 M KOH中以1.6 V vs RHE的电压下活化后的Sr3Fe2O7的HRTEM图(A)及其放大的晶格图(B)。虚线部分为非晶SrFeOOH层。(C)活化前后Sr3Fe2O7表面的Fe 2p、Sr 3d和O 1s XPS光谱。插图为Sr3Fe2O7活化前后的FTIR光谱。(D)核-壳结构Sr3Fe2O7@SrFeOOH的HAADF-STEM图和相应的EDS元素分布。包括Sr、Fe和O的分布以及它们在壳层中的混合分布。(E)Sr3Fe2O7@SrFeOOH在30和90 oC下的原位Fe 2p XPS谱。
HRTEM观察表明(图3A),Sr3Fe2O7纳米颗粒在电化学活化后表面重构出约5 nm厚度的非晶层(图3B)。XPS谱、HAADF-STEM图像和EDS元素分布(图3C, D)分析确定了Sr3Fe2O7表面非晶层是SrFeOOH,即Sr3Fe2O7在电化学活化后形成了Sr3Fe2O7@SrFeOOH核-壳复合结构。原位XPS分析显示,Sr3Fe2O7@SrFeOOH核-壳复合结构中Fe的结合能是温度依赖的,这表明抑制Sr3Fe2O7@中Fe电荷歧化将改变Sr3Fe2O7@SrFeOOH界面电荷传输特性。在升温过程中,Sr3Fe2O7@SrFeOOH电极的电化学活性面积(ECSAs)在TCD前后急剧提升(图4A插图),这表明热抑制电荷歧化增加了活性物种的数量。相应地,由ECSAs归一化后的LSV曲线(图4A)也表现出热敏感性。90 oC时,Sr3Fe2O7@SrFeOOH在450 mV过电位处的OER电流密度(J450)约为30 oC时J450的3500%(图4B),说明热抑制电荷歧化对OER具有明显的促进作用。而Sr3Fe2O7在90 oC时的体相电阻只比30 oC时的电阻值低了33%,这意味着如此高幅度OER提升不是由体相电阻下降引起的。因此,热抑制电荷歧化应是加速Sr3Fe2O7催化剂本体/SrFeOOH催化层界面处电子转移的主要原因,因为Fe4+(t2g3eg1)可以提供未占据的eg轨道用以转移电子而无需电子配对能。为了直观地监测热抑制电荷歧化对OER动力学的影响,我们计算了Sr3Fe2O7@SrFeOOH的OER活化能Q(图4C)。Sr3Fe2O7@SrFeOOH在TCD前后的Q突然下降表明催化材料的电子态发生了变化。热抑制电荷歧化导致Sr3Fe2O7的电子态发生了变化,从而降低了Sr3Fe2O7@SrFeOOH界面的电子转移能垒。原位UV-Vis吸收光谱(图4D)则很好地证明了Fe4+是SrFeOOH催化层的OER活性物种。
EIS谱可以反映热抑制电荷歧化对OER过程中电子转移的影响规律(图4E)。当T < TCD,谱信号横跨了低频(< 100 Hz,相角θL)和高频(> 100 Hz,相角θH)区域,这源自于催化层/电解液界面(θL)和催化剂本体/催化层界面(θH)的电子转移信号的耦合(图4E)。在OER电位下,θL的相位角明显小于θH,这可能意味着电子转移在本体催化剂/催化层界面处受阻。而θH值接近恒定,表明本体催化剂/催化层界面处的电子转移与温度无关。然而,当T > TCD时,θH随着温度的升高而急剧减小,表明热抑制电荷歧化加速了本体催化剂/催化层界面的电子转移。Vocp衰减则反映了双电层内电子转移的快慢(图4F)。Vocp衰减是由于电解液中活性物种对Feδ+(3<δ<4)的还原,并且衰减的速率依赖于温度,表明Sr3Fe2O7@SrFeOOH界面电子转移能垒是温度依赖的。

图4. 热抑制电荷歧化加速水氧化。(A)在扫描速率为10 mV/s、80% iR补偿条件下,ECSAs归一化的温度依赖LSV曲线。(B)随温度变化的电阻率下降率(红色柱)和在450 mV过电位下的电流密度(J450)的增长率(红色柱)。(C)温度倒数与交换电流密度对数的Arrhenius图。斜率表示OER活化能。(D)在90 oC的1M KOH中不同电位下Sr3Fe2O7@SrFeOOH的原位UV-Vis吸收光谱。(E)温度相关的Bode图。θL和θH表示低频相位角(< 100 Hz)和高频相位角(> 100 Hz)。(F)30、60、70、80和90 oC时,在10 mA cm-2条件下进行OER后监测的Vocp衰减。
根据以上结果可以发现,Sr3Fe2O7@SrFeOOH在OER过程中经历了两处界面电子转移:电子首先从OER中间体注入到SrFeOOH层,而后通过Sr3Fe2O7@SrFeOOH界面被提取到Sr3Fe2O7体相传至外电路。Sr3Fe2O7在T < TCD时存在Fe4+到Fe3+和Fe5+的电荷歧化转变,由于Fe3+的3d轨道被单电子完全占据,核-壳界面处的电子转移受到阻碍。当加热抑制电荷歧化以稳定Sr3Fe2O7中的Fe4+时,核-壳界面上的电子转移能垒下降。如图5所示,在Sr3Fe2O7的TCD前后,由于热诱导了材料电子态的转变,当完全抑制电荷歧化时,OER活化能突然降低,打破了反应速率-温度之间的线性Arrhenius关系。我们的发现明确证实了通过热场调节材料电子态促进电催化水分解策略的可行性,为克服水分解中的电子转移能垒开辟了新途径。

图5.(A)在氧化还原对介导的水氧化过程中,Fe基催化剂的电化学OER过电位来自催化层中高自旋Fe4+(t2g3eg1)到催化剂体相内高自旋Fe3+(t2g3eg2)的电子转移能垒。通过热耦合来抑制电荷歧化将为界面电子转移提供更多未占据的轨道。催化材料的电子态在没有热激励的情况下,热促进OER速率遵循线性的lnk - T-1 Arrhenius关系,即热扩散效应。但是,一旦在特定温度下触发材料热物理效应时,lnk - T-1的斜率发生变化,此时热可以调控催化剂的电子态,超越热扩散效应,显著提升OER性能。(B)OER过程中的界面电子转移过程。OER中间体(*OH, *O, *OOH)与活性中心*(SrFeOOH)在基本反应步骤中形成等能电子隧穿。qR是核坐标,qelectron是电子坐标。而在Sr3Fe2O7@SrFeOOH界面处的电子转移是通过Fe-O-Fe双交换作用在Fe4+(t2g3eg1)的未占据eg轨道中实现的。
热-电耦合的可再生能源制氢技术是从能量耦合增效的角度来提升电-氢转换效率,为可再生能源制氢提供独特的解决方案,为发展新一代高效可再生能源制氢技术提供了理论基础和材料设计方法。该工作于2023年12月27日以“Thermal suppression of charge disproportionation accelerates interface electron transfer of water electrolysis”为题发表在PNAS (2024 ,121 , e2316054120)上。37000cm威尼斯物理学院2020级博士生陆梦非为论文第一作者,37000cm威尼斯现代工程与应用科学学院闫世成教授为论文通讯作者,该研究得到了邹志刚院士和于涛教授的指导和帮助,获得了江苏省双碳专项资金和国家自然科学基金的经费资助。
关键词:热-电耦合;热抑制电荷歧化;能量场调控材料电子态;可再生能源制氢
论文信息:
Thermal suppression of charge disproportionation accelerates interface electron transfer of water electrolysis
Mengfei Lu, Yu Du, Shicheng Yan*, Tao Yu, Zhigang Zou
https://www.pnas.org/doi/10.1073/pnas.2316054120
 English
English


