电化学界面电子转移机制是电化学研究的重要课题。1931年,R. W. Gurney提出电化学反应电子转移量子力学机制(Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character, 1931, 134, 137-154),认为电极稳态电流起因于电极/电解液界面等能电子宏量隧穿效应。此后,1952年,W. F. Libby 提出了等能电子转移满足Franck-Condon原理 (J. Phy. Chem. 1952, 56, 863),即电子转移时间尺度远快于分子构型变化。而后,在1955-1965年间, R. A. Marcus 提出了Marcus电荷转移理论 (J. Chem. Phys. 1956, 24, 966; Ann. Rev. Phys. Chem. 1964, 15, 155;J. Chem. Phys. 1965, 43, 679), 明确指出在电化学反应过程中电子转移是绝热非辐射过程,并且电子转移时间尺度通常在10-15秒,而反应分子构型变化在10-13 -10-11秒,因此可以认为当电子转移发生时,反应物分子几何构型尚没有发生变化,电极与反应分子之间的电子转移遵循等能转移机制。
然而,目前在绝大多数的电化学反应机制描述中,均认为在催化中心电子转移遵循活性离子氧化-还原反应过程,为此,课题组以活性中心氧化和反应分子氧化级联反应为模型,通过EXAFS、Raman、UV-vis吸收、飞秒吸收等多种原位手段证实电化学反应发生时,高价态活性中心遵循未占据空轨道直接电荷转移机制,活性中心呈现类导体的电荷传输机制,不发生价态变化,并在此认知基础上,提出以分子中原子的双描述符局部软度值(dual local softness,∆sk)和最高占据分子轨道(HOMO)能级作为高效电氧化反应筛选判据。
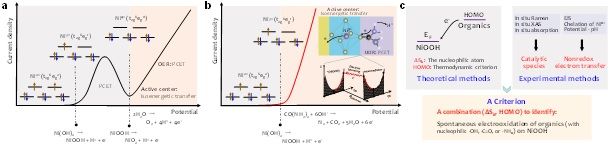
图1. 理解水氧化和尿素氧化机理性挑战。(a)碱性电解液中Ni(OH)2电极水电氧化反应的典型阳极极化曲线。(b)Ni(OH)2电极尿素电氧化反应的典型阳极极化曲线。(c)电荷转移机制研究所采用的理论和实验方法。
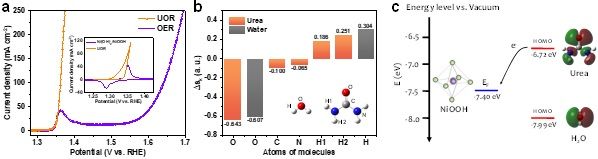
图2. 水氧化和尿素氧化的本质差异。(a)尿素氧化(UOR)和水氧化(OER)极化曲线对比。(b)以尿素和水分子中每个原子的∆sk识别亲核原子。(c)尿素和水的HOMO能级和NiOOH的费米能级对比。
为澄清电氧化催化层内电荷转移机制,首先对比了水氧化和有机物氧化二者差异性。对于Ni(OH)2电极,水氧化通常发生在Ni2+(OH)2/Ni3+OOH氧化之后,这意味着,尽管Ni2+ (3d8, t2g6eg2) 通过Ni(OH)2 → NiOOH + H+ + e- (质子耦合电子转移)氧化为Ni3+ (3d7, t2g6eg1),提供了可供电子转移的未占据轨道,但NiOOH仍然不能氧化水。相比之下,一旦Ni(OH)2被氧化成NiOOH,有机物(如醇、葡萄糖、尿素和联氨等)立即被电解。为理解Ni3+为什么能直接氧化尿素而不能氧化水,我们提出以(∆sk, HOMO)组合判据来进行理论解释,其中∆sk值的大小可以帮助判断分子中哪个原子是亲核位点,而HOMO能级与电极费米能级之间的关系可以作为反应能否发生的热力学判据。理论计算结果表明,NiOOH的费米能级(-7.4 eV)比尿素的HOMO能级(-6.72 eV)更负,说明尿素HOMO能级电子的化学势足够高,可以驱动电子从成键轨道向催化中心转移。并且∆sk值计算表明,在尿素分子中羰基氧是最亲核位点(∆sk = -0.643),能够发动亲核攻击Ni活性位建立能量转移通道。相比之下,虽然水分子中氧的∆sk也很负(-0.607),但水的HOMO能级(-7.99 eV)比NiOOH的费米能级更深,表明触发水氧化电子转移的热力学要求更高。这表明,这些有机物的电解反应的限速步骤为Ni(OH)2被氧化成NiOOH,即等能电子传输通道的建立。
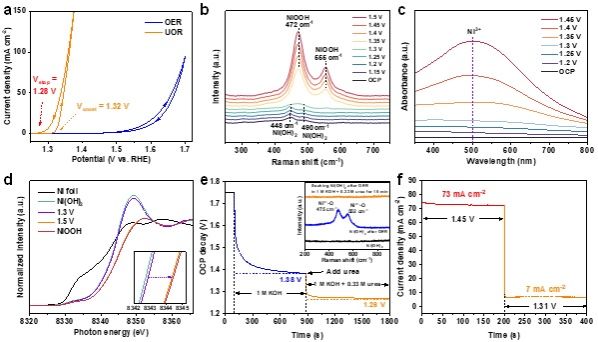
图3. 非氧化还原三价镍作为尿素电氧催化中心的证据。(a)计时电流-电位数据拟合得到的线性扫描伏安曲线。(b-d)Ni(OH)2电极尿素电氧化的电位依赖的b)原位拉曼光谱,c)原位紫外-可见吸收光谱,d)原位镍K边X射线吸收近边结构谱。(e)Ni(OH)2电极开路电压在电解液注入尿素前后的多段衰减。(f)当阳极电位在1.45 V和1.31 V之间阶跃时电流密度变化。
通过静态线性扫描伏安曲线、原位拉曼谱、原位紫外可见吸收谱、原位X射线近边吸收谱、开路电压衰减、快速切换电位等手段,我们发现三价镍氧化尿素是自发反应,且动力学过程极快。通过引入丁二酮肟耦合剂监测Ni2+离子(监测浓度极限nM,时间尺度ns),确认了Ni3+氧化尿素为亲核攻击和空轨道直接转移电子机制,这种非氧化还原的三价镍在氧化有机物中表现出优异的动力学特性。理论计算和实验证实,能够被三价镍高效氧化的有机物的HOMO能级在-7.4 eV到-6 eV(相对于真空能级)之间;并且其亲核官能团中亲核原子∆sk在-0.65到-0.15之间。有机物亲核官能团可以是羟基(甲醇、乙醇、苯甲醇)、羰基(甲酰胺、尿素、甲醛、葡萄糖和N-乙酰氨基葡萄糖)和氨基(苄胺)。快速的电氧化动力学可归因于有机物分子亲核位点和Ni3+ (t2g6eg1)未占据eg轨道的等能电子转移通道的建立。因此,我们可以清晰地描述完整的电化学电子转移过程,对于电催化氧化反应来说,外电压首先将活性中心从低价态极化到高价态,此过程可能伴随质子耦合电子转移的相变过程,处于高价态的催化活性中心与分子基元反应步物种通过轨道交叠形成成键轨道作为电子转移的能量通道,二者之间电子转移遵循Marcus电荷转移理论,即发生宏量等能电子隧穿,而催化中心则通过高价活性离子的未占据轨道直接转移电子,此时催化中心并不发生价态变化,通常遵循双/超交换机制转移电子至外电路。我们的发现对于设计高效催化剂和筛选动力学快速的有机电氧化具有重要借鉴价值。
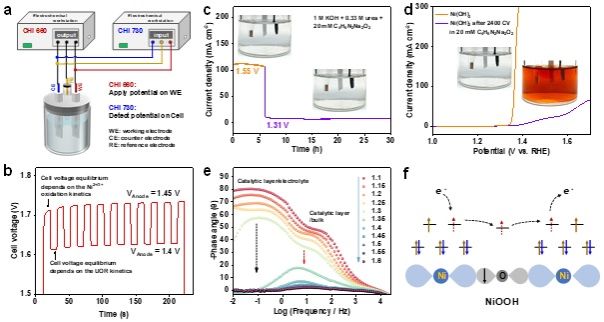
图4. 三价镍直接电荷转移动力学。(a)双化学工作站监测电极反应动力学。(b)当阳极电位在1.45 V和1.4 V之间阶跃时,电池反应动力学。(c)含有丁二酮肟钠盐的电解液中的恒电压反应及实时电解液光学照片。(d)含有丁二酮肟钠盐的电解液中CV循环扫模拟电子转移经由Ni(OH)2/NiOOH氧化-还原过程的电解液光学照片。(e)阻抗谱监测催化层/体相和催化层/电解液界面电子转移动力学。(f)通过Ni3+ (t2g6eg1)未占据的eg轨道进行直接电子转移示意图。
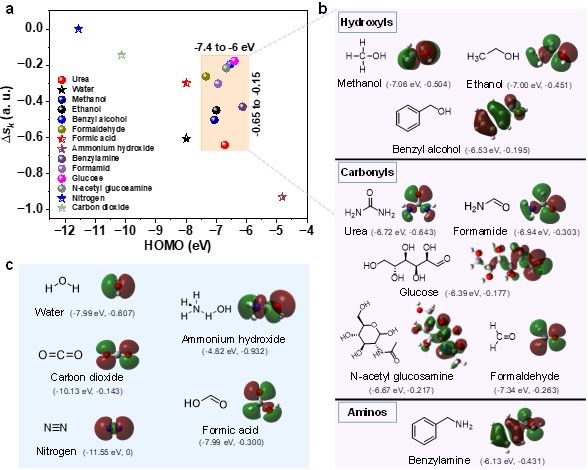
图5. 三价镍电氧化有机物的通用性。(a)以亲核原子∆sk和小分子的HOMO能级作为描述符,筛选Ni3+有效氧化的有机物。(b)可被Ni3+氧化的有机物HOMO能级和结构式。(c)不可被Ni3+氧化的有机物的HOMO能级和结构式。
该工作于2023年12月2日以“Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics”为题发表在Nature Communications 2023, 14, 7987上。37000cm威尼斯现代工程与应用科学学院博士生颜元东为论文第一作者、闫世成教授为论文通讯作者,该研究得到了邹志刚院士的精心指导,EXAFS测试得到了北京航空航天大学郝维昌教授的支持和帮助。研究得到了国家自然科学基金和江苏省双碳专项资金资助。
论文链接:
https://www.nature.com/articles/s41467-023-43649-6
DOI: https://doi.org/10.1038/s41467-023-43649-6
 English
English


